
Murasaki チュートリアル
Murasaki とは
Murasaki は 多種間のゲノム全体を高速にアライメントすることのできるツールで、 いくつかの新しいアイデアを導入することにより、 ゲノム再編成の全貌を高速に捉えつつも、 ゲノム間の細かな進化的関係も知ることができます。
Murasaki本体と、 その可視化ツールである GMVは、 それぞれオリジナルの開発元である、 慶應義塾大学理工学部 榊原研究室 のウェブサイトから入手が可能ですが、関係者が卒業したり、 GMV のコードを書いていた僕も異動してしまいましたので、 ダウンロードするだけで使うことのできるMac 用のバイナリは、 僕の研究室の プロジェクト > 生命科学 のページで配布を継続しています。現在配布中の Murasaki と GMV はどちらも、MacOS X 10.8 以降で動作するようになっています。
Murasaki のアイデア
ゲノム再編成
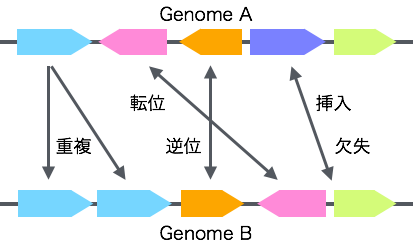
異なる生物種のゲノムを比べると、右の図のように、 ある領域は重複したり (重複)、 あるいは他の領域と順序が入れ替わったり (転移)、DNA のもう一方の鎖に移動したり (逆位)、 もともとのゲノムにはなかった配列が挿入されたり、 欠失したりしていることがあります。
これらはゲノム再編成とよばれ、 遺伝子の水辺伝播や相同組み換えや、トランスポゾンなど、 さまざまの要因によって起こります。 何らかの理由により頻繁に再編成が起きる生物のグループもあれば、 そうでないグループもあります。
種間や株間でゲノム全体の再編成を捉えることは、 その間の進化的関係を知ることになります。しかし、 ゲノム全体という大きなスケールで、 なおかつ何種類ものゲノムの間の関係を求めることは、 従来広く用いられている配列比較のためのツールでは、 計算時間や必要メモリなどの面で現実的ではありませんでした。
Murasaki では、 以下のいくつかのセクションで説明するようなアイデアを導入し、 ゲノムワイドの再編成を高速に計算することができます。
スペース入りシードパターン (Spaced Seed Pattern)
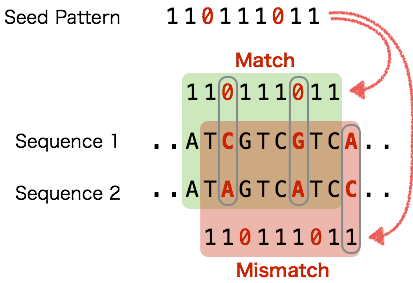
Murasaki でゲノム間の関係を調べる際には、 「シードパターン (あるいは単に、パターン)」と呼ばれる 1 と 0 で構成された文字列を指定します。たとえば、右の図では、 2本の短い配列に対して、"110111011" という長さ 9 のパターンを与えています。
これは、2本の配列の任意の9塩基の部分配列どうしを比べる際の、 match / mismatch の判定に使われます。 9塩基の部分配列どうしを比較する際に、パターンが "1" になっている位置では mismatch を許容せず、 "0" になっている位置での mismatch だけであれば部分配列全体としては match とする、というルールです。
右図の例では、10塩基の配列の中に3塩基の変異がありますが、 このルールと与えたパターンにより、左側の9塩基どうしは match、 右側の 9 塩基どうしは mismatch と判断します。
このように、シードパターンの長さと、 そこに含まれる 0 の位置や数で、 どのくらい厳密に部分配列間のマッチを行うかをコントロールできます。 基本的にはシードパターンが長くなるほど、 含まれる 0 の割合が少なくなるほど配列の比較は厳密になります。
また、シードパターンに含まれる 1 の数を、 そのパターンの「重さ (weight)」と呼んでいます。
シードとアンカー
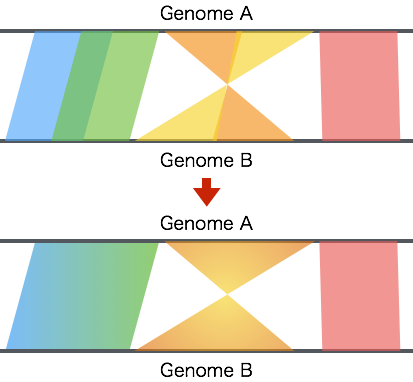
上で述べたように、対象の配列間で、 シードパターンの長さの部分配列 (seed) について match, mismatch を判定し、 (対象の配列が3本以上ある場合でも) すべての配列で match となった場合には、右図の上のように「アンカー (anchor)」 とよばれる線が引かれます。2つ以上のアンカーが同じ方向で、 かつ重なる・あるいは連続していた場合、右図の下のように、 お互いに結合され、ひとつの長いアンカーとなります。
アンカーの長さは、最短でシードパターンの長さ、 最長で入力として与えられた配列の全長になります (まったく同じ配列を2本与えれば、 配列全体がひとつのアンカーでカバーされます。)
アンカーが引かれた領域は基本的に、ある程度の相同性のある、 保存領域です。これはいくつもの遺伝子を含む 「シンテニー」と呼ばれる長さになることもあれば、 ひとつの遺伝子程度の長さであったり、 あるいは遺伝子の特定のドメインやコーディング領域程度の長さになることもあります。 これは、配列の相同性がどの程度あるか、ということや、 与えるシードパターンの長さや重さにもよって変わってきます。
tf-idf スコア
Murasaki では、シードパターンを用いて、 与えられた配列全てに共通に存在する部分配列を検出しますが、 この際に、ある部分配列が配列中に出現する頻度は、 部分配列によって異なります。
たとえば、 リピート配列のようにゲノム配列中に多数のコピーが存在する配列や、 その逆に、特定の遺伝子に固有の配列であれば、 1度しかゲノム中に出てきません。
ゲノム間の進化的な関係を読み解く上では多くの場合、 多数のコピーが存在する部分配列よりも、 コピー数の少ない部分配列に着目することが多いでしょう。 そこで、Murasaki では tf-idf (term frequency - inverse document frequency) とよばれる手法で、 アンカーのスコア付けを行っています。
これは、自然言語処理で、 文書を特徴付ける単語をスコアづけするのに用いられる基本的な手法で、 ある単語が特定の文書に出てくる回数 (tf: term frequency) に、 その単語を含む文書の数の逆数 (idf: inverse document frequency) をかけて算出します。
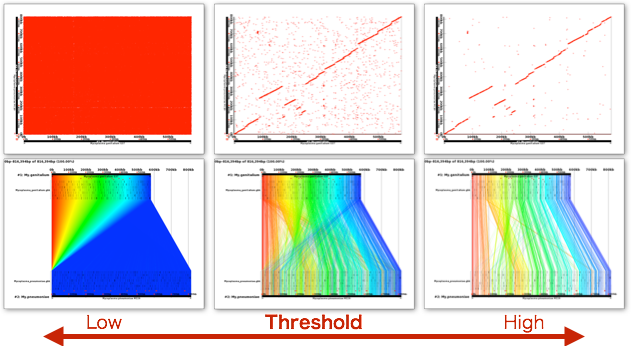
Murasaki では、seed を単語として数え、アンカーをスコア付けします。 右の図は、Murasaki の結果を tf-idf スコアの下限値でフィルタして並べたもので、 フィルタの下限値を引き上げるにつれてと、 ゲノム間で保存されている「骨格」 が浮かび上がってくることがわかります。
リピート耐性: mergefilter
Murasaki はゲノム配列全体をターゲットとして、 保存されている配列の検出を行いますが、 比較するゲノム間に共通のリピート配列が存在すると、 組み合わせが爆発することがあります。 従来はこういった問題を回避するために RepeatMasker などを使ってリピート配列をマスクしておく必要がありました。
Murasaki では、特定の seed の組み合わせが閾値を超えて多くなる場合に、mergefilter という機能を使うことで、 アンカーの構成要素から除外することができます。 これにより、事前にリピート配列に関して検討を行わなくても、 全ゲノムでの比較を高速に実行可能です
基本的な使い方
ここでは、MacOS X (10.8 以降) で Murasaki を実行し、 GMV を用いて 2 本のゲノム間のアライメントを俯瞰するところまでを解説します。
ソフトウェアと配列を手に入れる
Murasaki と GMV はどちらもこのウェブサイトの、 プロジェクト > 生命科学 のページから手に入れることができます。Murasaki 本体は実行ファイルがひとつだけで、 コマンドラインから実行しますので、zip ファイルを展開してできる murasaki という実行ファイルをどこか好きなフォルダに置いておきましょう (ここでは ~/bin/murasaki とします)。 GMV は GUI のアプリケーションですので、 これはデスクトップでも、アプリケーションフォルダでも、 どこでもかまいません。
チュートリアルで使用する配列は、 Mycoplasma genitalium G37と Mycoplasma pneumoniae M129とします。NCBI から Genbank 形式のファイルがダウンロードできますので、それぞれMy-genitalium.gbk、 My-pneumoniae.gbk という名前としておきます。 実際のファイル名は異なっていても、読み替えていただければOKです。
Murasaki を実行する
さきほどのふたつの配列ファイルが seq/ にあるとして、重さ 15、 長さ 21 の seed pattern で Murasaki を実行します。
% ~/bin/murasaki -p 15:21 -n genitalium-pneumoniae seq/My-genitalium.gbk seq/My-pneumoniae.gbk
Seed pattern は自分で -p 11001111 のように指定することもできますし、 この例のように重さと長さを指定してランダムに生成することもできます。 実際に使われた seed pattern は、実行開始直後に、
Writing to: output/genitalium-pneumoniae Pattern is: 101001110111111111001
のように表示されます。また、結果は output/ 以下に、 -n オプションで指定したファイル名に書き込まれます。
アンカーの計算は、
- 配列 1 の両方向の strand でハッシュを計算
- 配列 2 の両方向の strand でハッシュを計算
- アンカーを抽出
の順で行われます。ハッシュの計算というのは、配列に seed pattern を当ててスキャンしながら、どの seed がどの位置にあるか、 の一覧をつくる作業です。与えられた配列を両方向からスキャンし、 出現するすべての seed の一覧を作り終わったら、 アンカーを抽出します。
最終的に、
Anchor extraction completed in: 2.765 seconds Total anchors: 11251
などと表示されて計算が終了します。ここで、 もうすこし短くて軽い seed pattern、たとえば 12:18 を使うと、
Anchor extraction completed in: 16.381 seconds Total anchors: 414680
のように、アンカーの抽出にかかる時間も、 抽出されるアンカーの数もぐっと多くなります。 あまり多すぎると計算に時間がかかりすぎたりすることもありますので、 数字を見ながら少し長めの seed pattern から試してみるのがよいでしょう。 なお、このチュートリアルでは 15:21 や 12:18 のように指定して、 ランダムな seed pattern を使っているので、 実際にやってみたらアンカーの正確な数が違う、 ということもありますが、バグではありません。
結果のファイル
結果は、さきほどの説明の通り、output/ 以下に、-n オプションで指定したファイル名で書き込まれます。 いくつかのファイルが生成されますが、 いちばん基本的なものは .anchors ファイル (この例だと、 output/genitalium-pneumoniae.anchors) です。 このファイルには、以下のようにアンカーの位置が記録されています。
54 74 + 770742 770762 + 68 89 + -88583 -88562 - 69 90 + -23793 -23772 -
これは、My-genitalium.gbk の 54-74bp から My-pneumoniae.gbk の 770742-770762bp の同じ鎖に、68-89bp から 88583-88562bp の逆鎖に、 69-90bp から 23793-23872bp の逆鎖に、 それぞれアンカーが引かれているということを示しています。
また、それぞれのアンカーの tf-idf スコアは、.anchors.stats.tfidf ファイルに、
9.32821 9.32821 9.32821
のように記録されています。 さきほどの3つのアンカーがどれも同じ 9.32821 のスコアを持っていることがわかります。
そのほか、いくつかのファイルが生成されますが、 特に重要なところでは、 .options には実行時に指定したいろいろのパラメータや実際に使われた seed pattern が、.seqs には対象の配列ファイルの名前が、 それぞれ記録されています。必要に応じて参照してください。
GMV で結果を見る
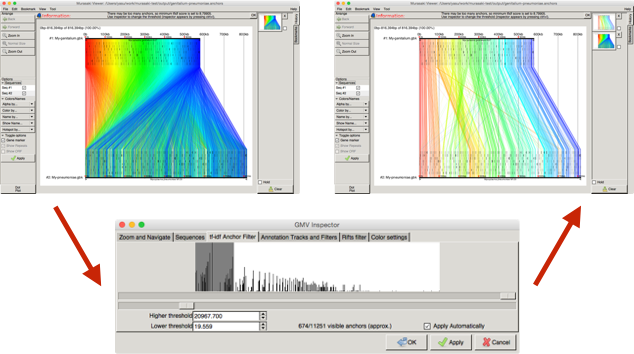
GMV を起動し、Murasaki の出力した .anchors ファイルを選択すると、その結果をグラフィック表示することができます。 アンカーが多い場合、 右図の左側のようになんだかわからなくなってしまいますが、 Inspector を開いて tf-idf anchor filter の下限値を引き上げると、 右図の右側のように、 ゲノムの再編成の様子が見やすくなります。
GMV では、一本一本の線がひとつひとつのアンカーに対応しており、 一番上に表示されているゲノム上での位置で線の色を決定しています。 したがって、上から2本目のゲノムで線の間隔が空いていれば、 そこには挿入された配列があることを意味していますし、 線が交差していれば、再編成が起きていることがわかります。 任意の場所を拡大したり、アンカーをたどったり、 その場所のアノテーションを参照したりと、 ゲノムブラウザ的な使い方をすることもできます。
GMV はたいへん多機能ですので、 また別の機会に紹介記事を書くことにします。
FAQ
3本以上の配列をアライメントできますか?
もちろんです。Murasaki ではたくさんの配列を与えた場合でも、 progressive にではなく一気にアライメントして、 すべての配列に共通に保存されている部分配列を探します。 リピート配列などがあった場合でも、mergefilter を適切に適用することで、計算の所要時間が突然爆発することを防ぎ、 高速な処理を実現しています。
また、"Rift" と呼ばれるオプションで、 いくつかの配列には含まれていない部分配列を、 計算結果のアンカーに含めることも可能です。
使える配列ファイルはなんですか?
基本的には GenBank 形式と、FASTA 形式を扱うことができます。 FASTA 形式の場合には、染色体や contig など、 複数の配列をひとつのファイルにまとめておき、 ひとつの長い配列として扱うことができます。この場合、 配列と配列の間には 10bp のギャップが挿入され、 配列の間をまたぐアンカーが生成されることはありません。
必要なメモリはどのくらいでしょうか?
Murasaki は EcoHash というきわめて省メモリな独自のデータ構造を採用しており、 具体的に 1bp あたり何バイトのメモリが必要、 ということを計算することはできませんが、 大腸菌くらいのサイズのゲノムを2種類比較するなら、 2GB 程度のメモリで充分です。最近のコンピュータはたいてい 8GB 以上のメモリがありますし、 数種類程度の微生物ゲノムならば問題なく扱うことができます。
また、Hash bits と呼ばれるパラメータ (-b オプション) の数字を小さめに調整することで、 少ないメモリでも計算が可能になることがあります。 この値は通常、空きメモリのサイズから自動的に計算されますが、 システムの状況によっては実際に確保可能なメモリより大きくなってしまうことがあります。使用された hash bits の値は .options ファイルに
Max usable hash bits: 27
のように記録されています。これを小さくすると、 より少ないメモリでの計算が可能ですが、計算にはより時間がかかります。 しかし、大きすぎる場合には、仮想メモリのスワップが発生し、 計算が終わらなくなる場合があります。
Seed pattern はどのように決めたらよいでしょうか
これは難しい質問ですが、大腸菌など小さなゲノムの場合には 15〜30bp くらいの長さ、ヒトなど大きなゲノムでは 100bp 以上の長さを指定することもあります。
もちろん、 特定のドメイン配列の長さなどに合わせて seed pattern を細かく決めたい、ということもあるかもしれませんが、 基本的には何度か Murasaki を走らせながら、try & error で長さと重さを決めていくのがよいと思います。
Murasaki を自分のシステムでコンパイルしたいのですが
基本的には gcc と Boost C++ Library があれば動作します。 LLVM Clang の C++ コンパイラではコンパイルできません。 これは、Murasaki に含まれている、Interval Tree というデータ構造の実装が強く GNU の STL 実装に依存しているためで、 残念ながらいまのところ解決の予定はありません。
Murasaki は並列システムで動作しますか?
OpenMPI などがあれば MPI による並列化に対応しています。 MIT-shmem があれば (最近の Unix 系の OS ではほとんど動くはず) 、ノード内での並列化も可能です。
並列化した場合には各ノードのメモリを集めて使うこともできるので、 大規模な解析を実行する場合には必須です。